Table Of Content

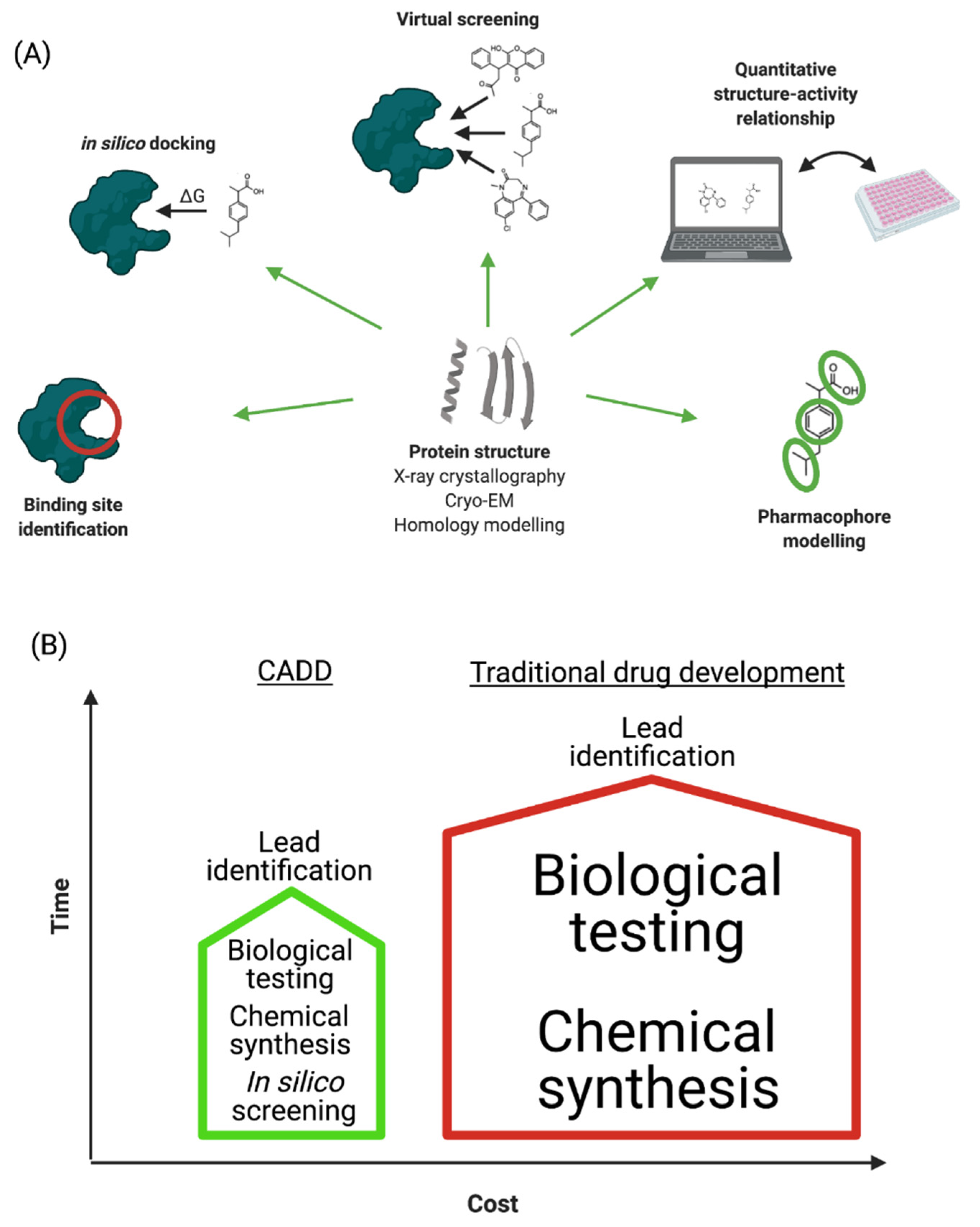
Databases should contain detail data on genomics and proteomics, high quality sequence information, physicochemical properties, and structures. In particular, like any computer assisted hypothetical system results must be validated in actual systems, and many lead molecules identified using CADD have failed to exhibit desired activities in biological systems [83, 84]. Several parameters must be met before potential compound to be approved as potent lead/drug, as it has to pass several pharmacological criteria. In fact, an average of only 40% of lead/drug candidates passes the different phases of clinical trials and obtains approval for clinical use. Computer-Aided Drug Design (CADD) emerged as an efficient means of identifying potential lead compounds and for aiding the developments of possible drugs for a wide range of diseases [8, 9].
5. Molecular Dynamic (MD) Simulation
Although the synthetic success rate for some of the commercial on-demand chemical spaces (for example, Enamine REAL Space) have been thoroughly validated20,21,22,23,24,26,42, synthetic accessibilities and success rates of other chemical spaces remain unpublished38. These are important metrics for the practical sustainability of on-demand synthesis because reduced success rates or unreasonable time and cost would diminish its advantage over custom synthesis. Al., the blue components are a part of computer-aided drug design (CADD) and it has transformed the way targets are being discovered and validated. In the modern era, the process filters active ingredients passing through different phases and choosing the best candidates to enter into clinical trials. We believe this review will be helpful for better understanding of CADD and its applications towards the discovery of new drug candidates against various fatal NDs. Straightaway the AI designed new molecules that also increase the activity of PPARs, like the drugs currently available, but without a lengthy discovery process.
Challenges of gigascale screening
When combined with more accurate in vitro testing, this may reduce and eventually eliminate animal test requirements (as recently indicated by FDA)145. The modular nature of on-demand virtual libraries supports further growth by the addition of reactions and building blocks. However, building, maintaining and searching fully enumerated chemical libraries comprising more than a few billion compounds become slow and impractical.
1. Structure-based Drug Design (SBDD)
His research encompasses discovery of neurological therapeutics, characterization of drug mechanism, and early and later stage drug development. Dr. Rogawski is an elected fellow of the American Association for the Advancement of Science and was awarded the UC Davis Chancellor's Innovator of the Year Award for inventing the drug Zulresso™. In addition to the common methods introduced in the first edition of this chapter, additional CADD methods developed recently in our laboratory as well as from other laboratories will be described below.
Global CADD Market Expected to Reach $7.81 Billion by 2030, Driven by Growing Demand for Advanced Drug Discovery - Yahoo Finance
Global CADD Market Expected to Reach $7.81 Billion by 2030, Driven by Growing Demand for Advanced Drug Discovery.
Posted: Fri, 29 Sep 2023 07:00:00 GMT [source]
Advanced computational applications have been shown to be effective tools and notable successes have been achieved using these techniques. CADD is a specialized discipline, whereby different computational methods are used to simulate interactions between receptors and drugs in order to determine binding affinities [19]. However, the technique is not limited to studies of chemical interactions and binding affinity predictions, as it has many more applications ranging from the design of compounds with desired physiochemical properties to the management of digital repositories of compounds.
Pharmacophore models mapping
The CADD methods presented in the chapter such as SILCS for SBDD or CSP for LBDD take this issue into account and thus have advantages over other CADD methods that only rely on single crystal structure or limited ligand conformations. CADD methods are mathematical tools to manipulate and quantify the properties of potential drug candidates as implemented in a number of programs. These include a range of publicly and commercially available software packages; the subset described below represents examples of fundamental tools for CADD with emphasis on those commonly used in our laboratory. Wet-lab, SBDD and LBDD CADD techniques are outlined in solid lines, dashed lines or dotted lines, respectively. Double headed arrows indicate the two techniques can be used interactively in several iterative rounds of ligand design. "Our work has made the world of proteins accessible for generative AI in drug research," Schneider says.
The SARS CoV-2 2′-o-methyltransferase (nsp16) is another important enzyme target essential for viral multiplication. The enzyme precisely protects the viral RNA from the cellular innate immunity by participating in the formation of a specific arrangement known as RNA cap, a structure which contributes to viral RNA stability and effective process of translation [124]. SARS-Cov-2 Nsp13 helicase is one of the critical enzyme among the 16 known CoV Nsp proteins which shows the highest sequence conservation across the CoV family, indicating their importance for viral multiplication.
Such gigascale virtual libraries are therefore usually maintained as non-enumerated chemical spaces, defined by a specific set of building blocks and reactions (or transforms), as comprehensively reviewed in ref. 38. Within pharma, one of the first published examples includes PGVL by Pfizer37,43, the most recent version of which uses a set of 1,244 reactions and in-house reagents to account for 1014 compounds. Other biopharma companies have their own virtual chemical spaces38,44, although their details are often not in the public domain. Among commercially available chemical spaces, GalaXi Space by WuXi (approximately 8 billion compounds), CHEMriya by Otava (11.8 billion compounds) and Enamine REAL Space (36 billion compounds)45 are among the largest and most established. In addition to their enormous sizes, these virtual spaces are highly novel and diverse, and have minimal overlap (less than 10%) between each other46.
What is drug discovery?
Several in silico experiments have been conducted using the modeled structure of cysteine protease to investigate the nature of its binding site. Protein-protein interactions (PPIs) are involved in a tremendous amount of vital cellular processes in bacteria and can serve as novel antibiotic targets (130, 131). Efforts towards the inhibition of PPIs related to division and replication, transcription, outer membrane protein complexes, as well as toxin-antitoxin systems in bacteria are ongoing (132). Thus, PPI prediction is of utility to identify novel druggable PPI interfaces in bacterial proteins and pave the way toward novel antibiotic therapeutics. Traditional PPI prediction methods are mostly based on rigid protein structures with limited flexibility considerations (133). Using the GFE FragMap and protein residue occupancy distributions, or protein probability grids (PPG), calculated from SILCS, a PPI prediction method named SILCS-PPI was put forward in our laboratory (134).
However, during the last decade, computers have been used to aid and accelerate the process of drug discovery, and this process is now referred to as computer-aided drug design (CADD) or computer-assisted molecular design (CAMD). Neurodegenerative disorders (NDs) are diverse group of disorders characterized by escalating loss of neurons (structural and functional). The development of potential therapeutics for NDs presents an important challenge, as traditional treatments are inefficient and usually are unable to stop or retard the process of neurodegeneration. Computer-Aided Drug Design (CADD) has emerged as an efficient means of developing candidate drugs for the treatment of many disease types.
LBDD is used in the absence of the target 3D structure with the central theme being the development of an SAR from which information on modification of the lead compound to improve activity can be obtained. Notably, CADD methods are evolving with researchers continually updating and implementing new CADD techniques with higher levels of accuracy and speed (24–26). In this chapter, we will present commonly used CADD approaches, including those used in our lab for the design of next-generation antibiotics.
This in silico process has achieved a position of great importance in the drug discovery field [21, 36-38]. Molecular docking has emerged over the last two decades and is now considered an indispensable tool for CADD and in the structural biology field, and has been shown to be more efficient than traditional drug discovery methods. Molecular docking has been greatly facilitated by dramatic growth in computer power and the increasing availability of small molecule and protein databases. Recent advancements in computer methods and access to 3D structural information of biological targets are set to increase the effectiveness of this technique and facilitate its large-scale application to studies of molecular interactions involved in ligand-protein binding.
Biomacromolecular therapeutics, or so called biologics, need to be carefully formulated to maximize protein stability and minimize viscosity, so as to ensure both efficacy and safety for highly concentrated formulations (141). Toward maximizing stability, biologics can be formulated with excipients to help minimize aggregation and denaturation of the biologic in a solution formulation (142). To assist the rational selection of excipients for biologics, we developed the SILCS-Biologics protocol (143, 144) which combines SILCS-PPI and SILCS-Hotspots as described above to predict both PPIs that can contribute to protein aggregation and increased viscosity, and binding sites of excipients.
This information is then combined to build a model for protein stability, aggregation and viscosity prediction. Further growth of the on-demand chemical space size and diversity is also supported by recent development of new robust reactions for the click-like assembly of building blocks. As well as ‘classical’ azide-alkyne cycloaddition click chemistry120, recognized by the 2022 Nobel Prize in chemistry121, and optimized click-like reactions including SuFEx122, more recent developments such as Ni-electrocatalysed doubly decarboxylative cross-coupling123 show promise. Other carbon–carbon forming reactions use methyliminodiacetic acid boronates for Csp2–Csp2 couplings124, and most recently tetramethyl N-methyliminodiacetic acid boronates125 for stereospecific Csp3–C bond formation. Each of these reactions applied iteratively can generate new on-demand chemical spaces of billions of diverse compounds operating with a limited number of building blocks. Similar to the routinely used automatic assembly of amino acids in peptide synthesis, fully automated processes could be carried out with robots capable of producing a library of drug-like compounds on demand using combinations of a few thousand diverse building blocks126,127,128.
For SBDD, protein 3D structure is required to explore atomic level details of the ligand-protein interactions. When no protein structure is available from the PDB, structure prediction methods such as homology modelling (92) were used traditionally to generate 3D models. With the surging of AI and related DL techniques, DL driven structure prediction methods such as RoseTTA fold (48) and AlphaFold (49) can now predict most protein 3D structures to a level of approaching experimental accuracy. In the recent challenging 14th Critical Assessment of protein Structure Prediction (CASP14), AlphaFold was demonstrated to greatly outperform other methods, and its predictions are competitive with experimental structures in a majority of cases. An updated view on different types of NDs, their effect on human population and a brief introduction to CADD, various approaches involved in this technique, ranging from structural-based to ligand-based drug design has been discussed.
However, with all MD based methods the user must perform careful analysis to assure that the conformational ensemble is adequately converged for effective use in CADD. 1Conformational flexibility of molecules is a very important feature no matter if it is a small ligand or a large protein. Thus conformational sampling of a protein or ligand that produces an ensemble of biological meaningful conformations is necessary either for SBDD or for LBDD.







No comments:
Post a Comment